アズレンのS2蛍光
はじめに
アズレンは5員環と7員環が縮環した非ベンゼン系芳香族化合物の代表例です.図1に示したとおり5員環は負電荷を,7員環は正電荷を帯びており,大きく分極しているために小さな共役系であるにも関わらず可視領域に吸収があります.
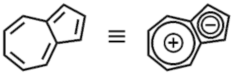
- 図1:アズレンの構造と分極
さらに興味深いことに,アズレンはS2状態から蛍光発光することが知られています.一般的な有機分子はKasha則として知られているとおり,最低励起状態(S1状態)から蛍光発光しますのでこれは特異な現象です.この原因は,アズレンのS1状態とS2状態のエネルギー差が異常に大きいためとされています.
本稿では,時間依存密度汎関数法(TD-DFT)によりこのエネルギー差について検証します.単にアズレンにおけるS1とS2のエネルギー差を求めてもわかりにくいので,構造異性体であるナフタレンについても計算を行い比較しました.
計算方法
今回の計算ではORCA 4.2.0を利用しました.計算はTD-DFT法で行い,汎関数にBHLYPを用いました.また,ORCAではハイブリッド汎関数にはTamm-Dancoff近似(TDA)がデフォルトで適用されますが,今回の計算では無効化しました.計算の高速化のためRIJCOSX近似を適用し,積分グリットにはGrid6,GridX8を指定しました.基底関数にはdef2-SVPを,補助基底関数にはdef2-SVP/Cを用いました.なお,BHLYP汎関数を用い,TDAなしのTD-DFTで計算をしないと収束が難しいケースがありました.
S2状態を計算する場合のインプットの例は以下になります.IRootのところを1にすればS1状態が,%tddftブロックを削除すれば基底状態(S0状態)計算になります.
! TightOpt NumFreq
! RIJCOSX BHLYP def2-SVP def2-SVP/C
! Grid6 NoFinalGrid GridX8
%tddft
NRoots 10
IRoot 2
MaxDim 8
Triplets false
TDA false
end
結果と考察
各状態の構造最適化を行い,平衡構造の全電子エネルギーを求めました.結果を表1に示します.なお,各状態間で結合長の多少の変化やそれに伴う結合角の変化はありましたが,基本的には平面のままで大きな構造変化は認められませんでした.このため,振動数計算によって求まるゼロ点振動を考慮しないでエネルギー差を比較しても問題ないと考えられます.
表1:アズレン,ナフタレンの各状態における全電子エネルギー
| 化合物 |
状態 |
全電子エネルギー [au] |
| アズレン |
S0 |
-385.314351303541 |
| S1 |
-385.233876682724 |
| S2 |
-385.175022513082 |
| ナフタレン |
S0 |
-385.376554310457 |
| S1 |
-385.211939607302 |
| S2 |
-385.203630705678 |
この表を元に各状態間のエネルギー差を求めます.このエネルギー差は各状態における構造緩和後のエネルギー差になります.なお,auとkcal/molの変換係数は627.5095です.
表2:アズレン,ナフタレンの各状態間のエネルギー差
| 化合物 |
遷移 |
エネルギー差 [kcal/mol] |
| アズレン |
S0→S1 |
50.5 |
| S1→S2 |
36.9 |
| ナフタレン |
S0→S1 |
103.3 |
| S1→S2 |
5.2 |
アズレンのS0→S1遷移のエネルギー差がナフタレンのそれの半分程度であるのは,可視光領域に吸収を持つことと対応しています.また,S1→S2遷移に着目すると,アズレンの状態間エネルギー差が36.9
kcal/molであるのに対し,ナフタレンのそれは5.2 kcal/molしかなく,アズレンはナフタレンに比べて約7倍のS1とS2間のエネルギー差を有していることが分かります.このような大きなエネルギーギャップのために,アズレンにおいてはS2からS1への緩和がスムーズに起こらず,S2から蛍光発光が起こると理解できます.
結論
TD-DFT計算によってアズレン,ナフタレンのS0, S1, S2状態のエネルギーを求め,そのエネルギー差を調べました.その結果,アズレンはナフタレンと比べ,約7倍のS1とS2間のエネルギーギャップ(36.9
kcal/mol)を有していることが分かりました.