結果と考察
まずは結合角の比較から入ります.ペンタン,3,3-ジフルオロペンタン,ジメチルシクロプロパンについて,構造最適化をDFT-TPSS-D3(BJ)/def2-TZVPPレベルで行いました.結果を図1に示します.
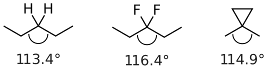
- 図1:ペンタン,3,3-ジフルオロペンタン,ジメチルシクロプロパンの結合角の比較
ペンタンでは中央の炭素周りの結合角が113.4°であったのに対し,ジメチルシクロプロパンでは114.9°になり,3,3-ジフルオロペンタンでは116.4°まで大きくなりました.結合角が置換基によって大きくなることがきちんと再現できています.結晶中の値よりプロパンは大きめ,他の2つは小さめの値となっていますが誤差の範囲でしょう.
では,このような結合角の変化が配座に与える影響はいかほどでしょうか.それを見積もるためanti配座とgauche配座で配座エネルギーを比較します.TPSS-D3(BJ)/def2-TZVPPレベルで構造最適化を行い,得られた構造を用いてLPNO-CEPA/1/def2-QZVPレベルでシングルポイント計算を行い,求まったエネルギーを比較することによって配座エネルギーを算出しました.結果を図2に示します.
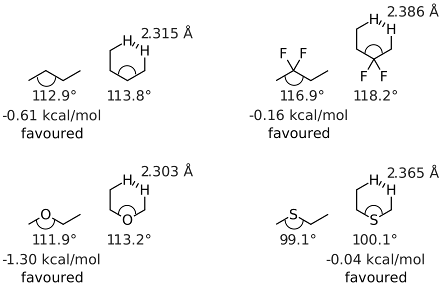
- 図2:ブタン,2,2-ジクロロブタン,エチルメチルエーテル,エチルメチルスルフィドのanti配座,gauche配座の構造と配座エネルギー
炭素だけのブタンは結合角が112.9°でしたが,フッ素置換では116.9°に大きくなりました.一方,エーテルやスルフィドの場合は.それぞれ111.9°99.1°にまで小さくなりました.これは,ローンペアは結合電子対よりも空間的に広がっているからと一般的に説明されています.一方,gauche配座では,ブタンは113.8°であったのに対し,フッ素置換では118.2°まで大きくなっています.一方,エーテルとスルフィドはanti配座と同様に炭素のみの場合よりも小さくなり,それぞれ113.2°,100.1°でした.
さらに,gauche配座の場合の最近接水素間距離を見ていくと,炭素のみでは2.315Åだったのが,エーテルでは2.303Åに縮まりました.一方,フッ素置換やスルフィドの場合は,それぞれ2.386Å,2.365Åまで伸びています.これは,フッ素置換の場合は結合角が広がっているため,スルフィドの場合にはS-C結合が長いためです.
それでは最後に配座エネルギーについてまとめます.まず,ブタンの場合はanti配座が0.61 kcal/mol有利です.フッ素置換になって結合角が大きくなると,立体障害も減ってエネルギー差が小さくなり,anti配座はわずかに0.16 kcal/molだけ有利という結果になりました.エーテルの場合はC-O結合がC-C結合よりも短いことが影響してか,anti配座が1.30 kcal/molも有利です.一方,スルフィドの場合はanti配座とgauche配座のエネルギー差は殆ど無い(gauche配座が0.04 kcal/molだけ有利)と言う結果になりました.おそらくS-C結合が長いために立体障害が小さくなっているからでしょう.